



















French Implant Scandal Dims Hope of Speedy US Device Approvals
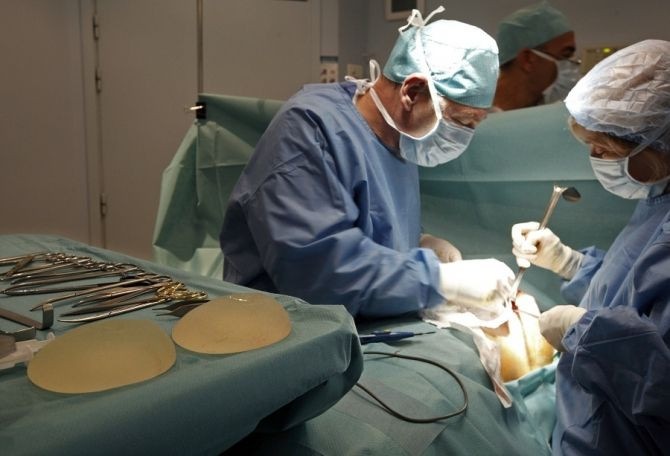
In the last year, medical device manufacturers have pressed U.S. officials to approve high-risk products quicker like their European counterparts, but the recent scandal over the thousands of faulty breast implants provided to women across Europe may dim the chances for a speedier approval process.
Last month French and German officials recommended thousands of women to have surgery to remove implants made by Poly Implants Protheses SA, after reports that the implants were leaking industrial silicone inside patients.
The leaky implants triggered more scrutiny over European rules for medical devices that doctors were already demanding for an overhaul.
Even though the implants were never approved for use in the U.S., the recent incidences could very well alter the debate for looser regulation by U.S. health officials. Congress will decide if U.S. device rules will be altered to follow Europe’s speedier model by September.
Although both congressional parties have supported the industry’s plea for a quicker approval method, recent cases of leaking implants and other EU device failures should be taken into consideration by lawmakers, according to device expert Carl Heneghan, an Oxford University professor.
“All the industry guys in the U.S. say, ‘we should have access to these products much sooner, like in Europe,’” Heneghan said to Bloomberg reporters. “The flip side is the European people are being used as guinea pigs.”
Heneghan is among the growing number of doctors, regulators and device makers calling for changes to the European system which depend on private reviewers hired by manufacturers to approve products, and the European Commission is expected to recommend changes by June of this year.
The PIP breast implant scandal is an inevitable result” of regulators’ “paralysis and inability to correct the failings of a flawed system,” said Richard Horton, the editor of The Lancet journal in an editorial released Wednesday. European approval “does not guarantee safety.”
U.S. device makers have long complained about slow reviews as well as inconsistent standards at the Food and Drug Administration. However, the FDA was recently reproached after some high-profile recalls it cleared for sale like the failure-prone Johnson & Johnson hip implants and Medtronic defibrillators that were likely to have lead fractures.
Last year industry supporters cited the EU as a better model, and said that the speedier review process was taking American jobs with them. In 2010, a Stanford University survey of manufacturers found complex devices reach the EU two years faster on average.
More than a dozen bills have been introduced to modify FDA rules.
“You can carry this business of safety to a ridiculous level,” said Representative Phil Gingrey, a Georgia Republican who sponsored one of the proposals said to Bloomberg “We’ve got to be responsible and safe, but we’ve got to understand that you can completely price these innovators out of the market.”
However, challengers of the European system said that regulatory authorizes have no control over how devices are actually made, and relied on “design specifications” to render approval decisions.
Patient safety expert Brian Toft said in a meeting with U.K. health minister Simon Burns in September 2011 that the CE Mark pathway was "a smokescreen for faulty and dangerous devices that place patients at risk."
Doctors at the European Society of Cardiology meeting in Paris last August said the European regulatory pathway was too lax when reviewing potentially dangerous products and said that the pathway uses the same procedures to review pacemakers as they do electric toasters.
"Where there have been isolated instances of devices that were associated with complications, those have disproportionately occurred in countries that have earlier approval – and that tends to be Europe," Cardiff University cardiologist Dr. Alan Fraser, who led a E.U. regulatory review panel at the ESC meeting, told reporters at the time.
“People expect to know about the products that are being put in their bodies,” John Brennan, regulatory-affairs director at device industry trade group Eucomed based in Brussels, said to Bloomberg. “We should have the equivalent transparency as the U.S.”
“The answer at the end of the day isn’t to scrap the U.S. standard of safety and effectiveness,” Jeffrey Shuren, director of the agency’s device-review center said to Bloomberg in response to an industry-sponsored study by the Boston Consulting Group that found little difference in the number or severity of recalls in both the U.S. and EU systems. “The answer is to keep the U.S. standard but let us do our job to improve the program.”







